
SINDROME DI BARDET-BIEDL
Codice esenzione : RN1380Definizione
Malattia autosomica recessiva caratterizzata da retinite pigmentosa; polidattilia, obesità, ritardo mentale, ipogenitalismo, displasia renale e bassa statura. La sindrome è stata distinta come entità separata rispetto alla sindrome di Laurence-Moon (From J Med Genet 1997 Feb;34(2):92-8) - Medline Thesaurus
Segni e sintomi
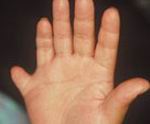
La sindrome è variabile nelle sue manifestazioni. Il 70% dei casi presenta una polidattilia post-assiale che, quando presente, può riguardare solo un arto.
E' stato riportato un caso con le caratteristiche della malattia che presentava una polidattilia pre-assiale (Manouvrier-Hanu et al. Bardet-Biedl syndrome with preaxial polydactyly; AM-J-MED-GENET 1999; 75-84).
La sindattilia e la brachidattilia sono molto comuni.
Una falange intermedia corta del 5° dito è di comune riscontro alla radiografia; anche un processo stiloideo ulnare corto o lungo, con i caratteri della deformità di Madelung, è presente nel 5-10% dei casi (Rudling et al. Skeletal abnormalities of hands and feet in Laurence-Moon-Bardet-Biedl syndrome: a radiographic study. SKELETAL-RADIOL 1996; 25: 655-660).
Gli eterozigoti risultano predisposti ad avere malformazioni renali e a sviluppare carcinoma renale. Problemi a carico dei reni sono molto comuni e possono essere presenti nel 90% dei casi, quando attentamente ricercati. Le anormalità includono allargamento dei calici renali, cisti o diverticoli, lobulazioni fetali, perdita di tessuto corticale ed una incapacità a concentrare le urine; si può instaurare insufficienza renale. Può essere presente anche displasia renale. L'aspetto può essere quello di una nefronoftisi giovanile. Nelle femmine sono molto comuni anomalie a carico dell'apparato urogenitale quali: atresia vaginale, vagina settata, ectopie dell'uretra, ipoplasia o duplicazioni dell'utero e delle tube di Falloppio.
Morbo di Hirschsprung ed atresia anale sono associati raramente alla malattia. Occasionalmente è stata riscontrata inoltre la presenza di situs viscerum inversus. Nei pazienti con sindrome di Bardet-Biedl l'ecocardiografia può rilevare alcune anomalie cardiache come ad esempio: cardiomiopatia dilatativa, valvola aortica bicuspide, ispessimento del setto interventricolare, stenosi della valvola polmonare (Elbedour et al. Cardiac abnormalities in the Bardet-Biedl syndrome: echocardiographic studies of 22 patients AM-J-MED-GENET 1994;52: 164-169). Il ritardo mentale non è sempre presente. Altre manifestazioni della malattia sono obesità (90%), diabete mellito(50%), ipogonadismo nei maschi (88%) ed irregolarità del ciclo mestruale nelle femmine(100%). Le anomalie a carico dell'apparato visivo consistono caratteristicamente in una retinite pigmentosa atipica con interessamento precoce della macula. Gli studi elettrofisiologici rivelano la presenza di una degenerazione dei coni e dei bastoncelli. L'insorgenza dei deficit visivi avviene nella seconda e terza decade. Altre condizioni di comune rilievo sono: miopia, astigmatismo, nistagmo, glaucoma e cataratta (R.M. Winter, M. Baraitser, London Dysmorphology Database, Oxford Medical Databases, 2000).
Recentemente sono state identificatre altre manifestazioni cliniche della malattia che includono deficit neurologici, del linguaggio, caratteristiche comportamentali, dismorfismo facciale, anomalie dei denti (Beales et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey J-MED-GENET 1999; 36(6):437-446).
Storia naturale
L'età media alla quale si giunge ad una corretta diagnosi è 9 anni; la diagnosi avviene tardivamente considerando che tale condizione è altamente debilitante, ma probabilmente questo è dovuto al lento instaurarsi delle manifestazioni cliniche. La polidattilia postassiale è presente nel 69% dei pazienti alla nascita, ma l'obesità si manifesta attorno ai 2-3 anni di età e la degenerazione retinica non è evidenziabile in media fino agli 8,5 anni di età (Beales et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey J-MED-GENET 1999; 36(6): 437-46). Circa il 25% dei pazienti sviluppa insufficienza renale entro i 48 anni di età, ma possono essere evidenziate anomalie con tecniche di imaging nel 96% dei pazienti (O'Dea et al. The importance of renal impairment in the natural history of Bardet-Biedl syndrome AM-J-KIDNEY-DIS 1996;27:776-783).
Nella seconda e terza decade è comune l'insorgenza di deficit visivi (Fulton et al. Natural course of visual functions in the Bardet-Biedl syndrome ARCH-OPHTALMOL 1993;111:1500-1506).
L'età media alla quale si verifica il decesso è di 43 anni nei maschi e 46,4 nelle femmine. Le causedi morte sono principalmente insufficienza renale o infarto del miocardio (Riise et al. The cause of death in Laurence-Moon Bardet-Biedl ACTA OPHTHAL SCAN 1996;74:45-47).
Eziologia
La malattia è trasmessa come tratto autosomico recessivo; fino ad ora sono stati identificati 5 diversi loci: 2q31; 3p11-3p13; 11q13; 15q22; 16q13-16q22. Alleli presenti in loci differenti possono determinare l'espressione di fenotipi leggermente diversi. I casi associati a mutazioni sul cromosoma 3 presentano polidattilia a tutti e quattro gli arti, mentre i casi associati al cromosoma 15 presentano polidattilia che riguarda principalmente le mani. I casi associati al cromosoma 16 sono meno inclini a sviluppare obesità, mentre nei casi associati al cromosoma 15 essa ha un'insorgenza precoce (R.M. Winter, M. Baraitser, London Dysmorphology Database, Oxford Medical Databases, 2000). I pazienti BBS1 (locus11q13) tendono ad essere più alti dei loro genitori ed inoltre richiedono un sostegno scolastico minore rispetto ai pazienti BBS4 (locus15q22) (Beales et al. Bardet-Biedl syndrome: a molecular and phenotype study of 18 families J-MED-GENET 1997; 34:92-98).
Diagnosi
In generale per la diagnosi devono essere presenti quattro delle cinque manifestazioni principali della sindrome (ritardo mentale, retinite pigmentosa, polidattilia, obesità, ipogenitalismo) (R.M. Winter, M. Baraitser, London Dysmorphology Database, Oxford Medical Databases, 2000).
È necessario un approccio diagnostico multidisciplinare: pediatrico, oculistico, nefrologico; inoltre i pazienti affetti da questa sindrome dovrebbero essere sottoposti ad un programma diagnostico cardiologico per rilevare malattie dell'apparato cardiovascolare congenite od acquisite che potrebbero rientrare, secondo la letteratura, tra le manifestazioni della sindrome (Suilmann et al. Bardet-Biedl-syndrome. Is there a cardiac manifestation? HERZ-KREISL. 1997; 29/4 (121-124).
La sindrome di Bardet-Biedl è distinta dalla sindrome di Laurence-Moon. La differenza consiste nel fatto che i pazienti con sindrome di Laurence-Moon presentano spasticità e non sono affetti da obesità e polidattilia. Entrambe le condizioni si presentano con degenerazione retinica, ritardo mentale e ipogenitalismo (R.M. Winter, M. Baraitser, London Dysmorphology Database, Oxford Medical Databases, 2000). La sindrome di Bardet-Biedl ha dei segni clinici comuni anche ad altre sindromi, ad esempio la sindrome di Alstrome. Gli esami oftalmologici ed elettrofisiologici sono indispensabili per una corretta diagnosi dela sindrome di Bardet-Biedl (Ingster-Moati et al. Functional visual explorations of Bardet-Biedl syndrome: a study of three cases JOURNAL- FRANCAIS-D-OPHTALMOLOGIE 2000; 23(8):802-808). Metrocolpocele e polidattilia postassiale sono presenti sia nella sindrome di Bardet-Biedl che nella sindrome di McKusick-Kaufman; tuttavia, anomalie dell'utero e delle tube di Falloppio sono frequenti nelle femmine affette da sindrome di Bardet-Biedl e sono utili ai fini della diagnosi differenziale (Slavotinek et al. Phenotypic overlap of McKusick-Kaufman syndrome with Bardet-Biedl syndrome: A literature review AM-J-MED-GENET 2000; 95 (3): 208-215).
Diagnosi prenatale e prevenzione
Non è attualmente disponibile. È comunque possibile effettuare la diagnosi prenatale di polidattila mediante l'ecografia, ma questa manifestazione clinica non è presente in tutti i pazienti con sindrome di Bardet-Biedl (Zimmer et al. Fetal polydactyly diagnosis during early pregnancy: Clinical applications AMERICAN-JOURNAL-OF-OBSTETRICS-AND-GYNECOLOGY. SEP 2000; 183 (3): 755-758).
Terapia
Il trattamento è sintomatico. Esso comprende il trattamento dell'obesità e delle sue complicanze, dell'ipogonadismo, dei deficit visivi, dell'insufficienza renale, quando presente (Orphanet). Possono essere necessari interventi chirurgici per la ricostruzione dei genitali (Bruhl et al. Bardet-Biedl syndrome - aspects of nephro-urology and human genetics KLINISCHE-PADIATRIE JAN-FEB 2001; 213 (1): 8-12).
Bibliografia
Bruhl-P; Schwanitz-G; Mallmann-R; Muller-SC; Raff-R
Bardet-Biedl syndrome - aspects of nephro-urology and human genetics.
KLINISCHE-PADIATRIE. JAN-FEB 2001; 213 (1) : 8-12
Oeffner-F; Bornholdt-D; Ziegler-A; Hinney-A; Gorg-T; Gerber-G; Goldschmidt-HP; Siegfried-W; Wright-A; Hebebrand-J; Grzeschik-KH
Significant association between a silent polymorphism in the neuromedin B gene and body weight in German children and adolescents
ACTA-DIABETOLOGICA. JUN 2000; 37 (2) : 93-101
Beales-PL; Reid-HAS; Griffiths-MH; Maher-ER; Flinter-FA; Woolf-AS
Renal cancer and malformations in relatives of patients with Bardet-Biedl syndrome
NEPHROLOGY-DIALYSIS-TRANSPLANTATION. DEC 2000; 15 (12) : 1977-1985
Beales-PL; Reid-HAS; Griffiths-MH; Maher-ER; Flinter-FA; Woolf-AS
Renal cancer and malformations in relatives of patients with Bardet-Biedl syndrome
NEPHROLOGY-DIALYSIS-TRANSPLANTATION. DEC 2000; 15 (12) : 1977-1985
Ingster-Moati-I; Rigaudiere-F; Choltus-de-Petigny-MC; Bremond-Gignac-D; Lestrade-C; Grall-Y
Functional visual explorations of Bardet-Biedl syndrome: a study of three cases
JOURNAL-FRANCAIS-D-OPHTALMOLOGIE. OCT 2000; 23 (8) : 802-808
Slavotinek-AM; Biesecker-LG
Phenotypic overlap of McKusick-Kaufman syndrome with Bardet-Biedl syndrome: A literature review
AMERICAN-JOURNAL-OF-MEDICAL-GENETICS. NOV 27 2000; 95 (3) : 208-215
Chen-BH; Chiou-SS; Tsai-RK; Lin-YF; Wu-JR
Acute lymphoblastic leukemia in one of two siblings with Alstrom syndrome
JOURNAL-OF-THE-FORMOSAN-MEDICAL-ASSOCIATION. OCT 2000; 99 (10) : 792-795
Mishriki-YY
Can you name this congenital syndrome? Bardet-Biedl syndrome
POSTGRADUATE-MEDICINE. OCT 2000; 108 (5) : 207-209
Ghadami-M; Tomita-HA; Najafi-MT; Damavandi-E; Farahvash-MS; Yamada-K; Majidzadeh-A-K; Niikawa-N
Bardet-Biedl syndrome type 3 in an Iranian family: Clinical study and confirmation of disease localization
AMERICAN-JOURNAL-OF-MEDICAL-GENETICS. OCT 23 2000; 94 (5) : 433-437
Zimmer-EZ; Bronshtein-M
Fetal polydactyly diagnosis during early pregnancy: Clinical applications
AMERICAN-JOURNAL-OF-OBSTETRICS-AND-GYNECOLOGY. SEP 2000; 183 (3) : 755-758
Slavotinek-AM; Stone-EM; Mykytyn-K; Heckenlively-JR; Green-JS; Heon-E; Musarella-MA; Parfrey-PS; Sheffield-VC; Biesecker-LG
Mutations in MKKS cause Bardet-Biedl syndrome
NATURE-GENETICS. SEP 2000; 26 (1) : 15-16
Katsanis-N; Beales-PL; Woods-MO; Lewis-RA; Green-JS; Parfrey-PS; Ansley-SJ; Davidson-WS; Lupski-JR
Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome
NATURE-GENETICS. SEP 2000; 26 (1) : 67-70
Levy-M; Feingold-J
Estimating prevalence in single-gene kidney diseases progressing to renal failure
KIDNEY-INTERNATIONAL. SEP 2000; 58 (3) : 925-943
Seidemann-K; Lauten-M; Gappa-M; Offner-G; Latta-K; Ehrich-JHH
Obstructive airway disease caused by Moraxella catarrhalis after renal transplantation
PEDIATRIC-NEPHROLOGY. AUG 2000; 14 (8-9) : 707-709
Tomas-Zuber-M; Mary-JL; Lesslauer-W
Control sites of ribosomal S6 kinase B and persistent activation through tumor necrosis factor
JOURNAL-OF-BIOLOGICAL-CHEMISTRY. AUG 4 2000; 275 (31) : 23549-23558
Farooqi-IS; O'-Rahilly-S
Recent advances in the genetics of severe childhood obesity
ARCHIVES-OF-DISEASE-IN-CHILDHOOD. JUL 2000; 83 (1) : 31-34
Hrynchak-PK
Bardet-Biedl syndrome.
Optom-Vis-Sci. 2000 May; 77(5): 236-43
Beales-PL; Elcioglu-N; Woolf-AS; Parker-D; Flinter-FA
New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey.
J-Med-Genet. 1999 Jun; 36(6): 437-46
Fonte: http://malattierare.regione.veneto.it/default.htm
Nessun commento:
Posta un commento